Marjo S. van der Knaap (род. в 1958г.)
Син.: мегалэнцефалическая лейкоэнцефалопатия с субкортикальными кистами, MLC (англ.). Наследственное (АР) демиелинизирующее заболевание ЦНС из группы лейкодистрофий с дебютом в возрасте от рождения до 25 лет (преимущественно в 6 месяцев), характеризующееся диффузной субкортикальной лейкоэнцефалопатией с кистозной дегенерацией белого вещества. Заболеваемость и распространённость точно не известны, всего в специальной литературе описаны более 150 случаев. Заболевание преобладает в популяциях с кровнородственными браками и наиболее распространено среди касты агравал в Восточной Индии (штат Раджастхан), все пациенты в которой имеют один и тот же же биаллельный патогенный вариант (c.135dupC), что указывает на «эффект основателя», и ливийских евреев. В основе заболевания лежит гомозиготная/сложная гетерозиготная мутация гена MLC1 (локус 22q13) – классический вариант (76% случаев), или мутации гена HEPACAM (локус 11q24) – гомозиготная/сложная гетерозиготная (вариант MLC2A) или гетерозиготная (вариант MLC2В: ремиттирующая мегалэнцефалическая лейкоэнцефалопатия с субкортикальными кистами с умственной отсталостью или без неё, наследуется по АД-типу с неполной пенетрантностью, при этом часто наблюдаются мутации de novo). Ген MLC1 состоит из 12 экзонов и кодирует трансмембранный белок, связывающийся с β1-субъединицей Na,K-АТФазы в мультипротеиновом комплексе, регулируя тем самым реакцию Na,K-АТФазного комплекса на осмотический стресс. Ген HEPACAM состоит из 7 экзонов и отвечат за синтез молекулы клеточной адгезии семейства иммуноглобулинов, которая экспрессируется преимущественно в ЦНС (по крайней мере, мутации этого гена отражаются именно на ЦНС). Экспрессия обоих генов происходит в окончаниях астроцитов и межастроцитарных соединениях в ЦНС.
У 5% пациентов с энцефалопатией Ван дер Кнаап не удаётся обнаружить ни одного из вышеперечисленных генетических дефектов.
Уже при рождении (а чаще, на первом году жизни) становится заметной макроцефалия, которая присутствует практически у всех пациентов. После года рост окружности головы замедляется и вскоре размеры её начинают приближаться к норме. Вначале умственное и физическое развитие как правило соответствуют возрастной норме, однако, постепенно становится заметным несущественное отставание (умственное развивается позже и протекает мягче физического).

Внешний вид ребёнка с энцефалопатией Ван дер Кнаап (источник: Abdel-Salam G.M.H., Abdel-Hamid M.S., Ismail S.I. et al. Megalencephalic leukoencephalopathy with cysts in twelve Egyptian patients: novel mutations in MLC1 and HEPACAM and a founder effect // Metab. Brain Dis., 2016. – Vol.31. – P.1171-1179)
Внешний вид ребёнка с энцефалопатией Ван дер Кнаап (источник: Bhattacharyya K.B., Rai S. Megaencephalic leukoencephalopathy with subcortical cysts in a young Bengali girl // Neurol. India, 2015. – Vol.63. – P.436-743)
Когда ребёнок начинает ходить, становятся заметными нарушения походки – она неуверенная, дети часто падают. Мышечный тонус при этом имеет тенденцию к понижению, за исключением тонуса стоп, который обычно повышен. В первые годы жизни отмечается медленное прогрессирование нарушений моторики, однако, на первый план постепенно начинает выходить статическая и динамическая атаксия, атаксия походки. Также постепенно присоединяется дизартрия, иногда – с дисфагией. Появляются пирамидные знаки, спастичность, реже – экстрапирамидные нарушения (дистония, атетоз, тики). Способность к самостоятельному передвижению постепенно утрачивается, и к 10-20 годам многие пациенты становятся прикованы к инвалидной коляске; в более благоприятных случаях она сохраняется до 50 лет.
Иногда возникают поведенческие расстройства, аутизм. Очень характерны эпилептические приступы, развивающиеся на первом году жизни; обычно они хорошо поддаются лечению, но нередки (15-20%) эпилептические статусы, как правило, развивающиеся в течение нескольких лет после дебюта эпилептического синдрома. Даже малейшая травма головы может приводить к временному нарастанию клинических проявлений, в особо тяжёлых случаях – вплоть эпилептического статуса или до коматозного состояния.
Выделяют 2 разновидности фенотипической картины заболевания – классическую (варианты MLC1 и MLC2A) и благоприятную (вариант MLC2B). По сравнению с вышеописанными проявлениями классического фенотипа, благоприятный вариант характеризуется более мягкой манифестацией и улучшением двигательных функций после года жизни, более редким развитием эпилептического синдрома (лишь у 10% пациентов).
У родителей пробанда – носителей мутаттного гена – может наблюдаться макроцефалия без нарушений двигательных и когнитивных функций, в некоторых случаях – когнитивные/поведенческие расстройства и неловкость движений.
Типичными МРТ-признаками классической формы являются: 1) мегалэнцефалия; 2) диффузное двухстороннее симметричное поражение полушарного белого вещества (гиперинтенсивное в режиме Т2 и гипоинтенсивное в режимеТ1) с признаками его незначительного отёка; 3) относительная сохранность центральных структур (мозолистого тела, внутренней капсулы, ствола мозга); 4) относительно несущественное поражение белого вещества мозжечка без признаков отёка; 5) практически патогномоничное наличие субкортикальных кист в передних отделах височных долей, а также зачастую – и лобно-теменных областях; 6) регресс отёчности белого вещества со временем, приводящий к появлению картины атрофии мозга; 7) повышенная проницаемость отёчного белого вещества в режиме DWI (диффузионно-взвешенных изображений).
В некоторых случаях субкортикальные кисты могут увеличиваться в размерах и количестве. Иногда они достигают больших размеров, распространяясь на значительные объёмы белого вещества лобно-теменных областей. Возможен также и регресс патологических изменений белого вещества.
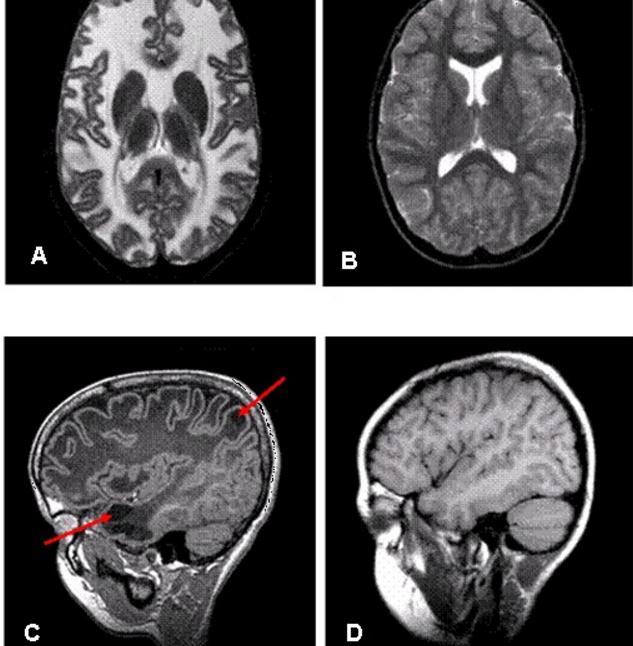
МРТ-картина энцефалопатии Ван дер Кнаап (рис. А,С) и нормального головного мозга (рис.B, D). А – Т2-взвешенное поперечное изображение головного мозга 9-летнего ребёнка с энцефалопатией Ван дер Кнаап: видно диффузное поражение белого вещества полушарий с признаками его отёка; С – Т2-взвешенное сагиттальное изображение головного мозга того же ребёнка, на котором видны субкортикальные кисты в передневисочной и теменной областях (показаны стрелками) (источник: Van der Knaap MS., Abbink T.E.M., Min R. Megalencephalic Leukoencephalopathy with Subcortical Cysts. Synonym: Van der Knaap Disease. – in: Adam M.P., Ardinger H.H., Pagon R.A. et al., eds. Gene Reviews // Seattle (WA): University of Washington, Seattle; 1993-2018)
МРТ-картина благоприятной формы энцефалопатии Ван дер Кнаап на первом году не отличается от таковой при классической форме (за исключением разве что практически интактного белого вещества мозжечка); в последующем наблюдается существенный регресс патологических изменений – через несколько лет головной мозг может выглядеть практически нормальным, с минимальным поражением белого вещества лобной и теменной областей и сохранившимися субкортикальными кистами в передневисочных областях.
Верификацию диагноза можно осуществить методом секвенирования – вначале производится секвенирование гена MLC1, затем, при отрицательном результате – гена HEPACAM (при благоприятном варианте вначале проводится анализ гена HEPACAM). Если патогенетический вариант не найден, возможно применение метода поиска делеции/дупликации в этих генах.
Описаны случаи повышения уровня глицина в ликворе {Sener}.
Дифференциальный диагноз проводится с рядом заболеваний, ядром клинической картины которых является макроцефалия с диффузной лейкоэнцефалопатией: болезнью Канаван(–Ван-Богарта – Бертрана), болезнью Александера, GM2-ганглиозидозами (I типа – болезнь Тея – Сакса, II типа – болезнь Зандхоффа(–Яцкевича(–Пильца)), GM1-ганглиозидозами (ранний и поздний инфантильные типы), L-2-оксиглутаровой ацидурией. Кроме того, некоторые случаи мерозин-дефицитной врождённой мышечной дистрофии типа 1А могут протекать с макроцефалией. Детали клинической картины и течение этих заболеваний, как правило, всё же имеют отличия; МРТ-картина ни одного из них не включает все признаки энцефалопатии Ван дер Кнаап. Если к году жизни окружность головы больного ребёнка существенно превышает норму, то с весьма высокой вероятностью у него нет энцефалопатии Ван дер Кнаап.
Прогноз для жизни при этом заболевании в целом благоприятный, хотя описаны случаи смерти в возрасте 10-20 лет. Если пациент сохраняет способность к ходьбе, самостоятельной или с поддержкой, к 15 годам, то скорее всего он будет оставаться ходячим и в последующем.
Патогенетического лечения до настоящего времени не разработано. Применение таких препаратов, как диуретики, ацетазоламид, креатинина моногидрат не продемонстрировало эффективности. Показана физиотерапия, логопедическая помощь, обучение по индивидуальному плану, антиконвульсанты при развитии эписиндрома. Учитывая высокий риск осложнений при травмах головы в потенциально травматичных ситуациях рекомендуется ношение шлема.
Впервые описана индийским неврологом Бхим Сен Сингхалом (род. в 1933г.) в 1991г. (Singhal B.S., Gursahani R.D., Biniwale A.A., Udani V.P. Tokyo, Japan: In Proceedings of the 8th Asian and Oceanian Congress of Neurology; 1991. Megalencephalic leukodystrophy in India; p.72). Однако, более подробное клинико-радиологическое описание этой нозологической формы было проведено позже, в 1995г., голландским детским неврологом Марьо Ван дер Кнаап (van der Knaap M.S., Barth P.G., Stroink H., van Nieuwenhuizen O., Arts W.F., Hoogenraad F., Valk J. Leukoencephalopathy with swelling and a discrepantly mild clinical course in eight children // Annals of neurology, 1995. – Vol.37. – N.3. – P.324-334), именем которой она и была названа, хотя сама М.Ван дер Кнаап не считала это нужным (всего ей было описано 5 нозологических форм).