Jean Alfred Émile Chavany (1892-1959)
Син.: (передний) оперкулярный синдром.
Крайне редко встречающаяся уникальная разновидность коркового надъядерного (псевдобульбарного) паралича, обусловленного двухсторонним (гораздо реже – односторонним) поражением (чаще всего, вследствие инфаркта) лобно-теменной покрышки (operculum frontoparietale) – участка коры большого мозга, в который входят задняя часть нижней лобной извилины, нижние отделы пре- и постцентральной извилин и нижний отдел передней части теменной доли.
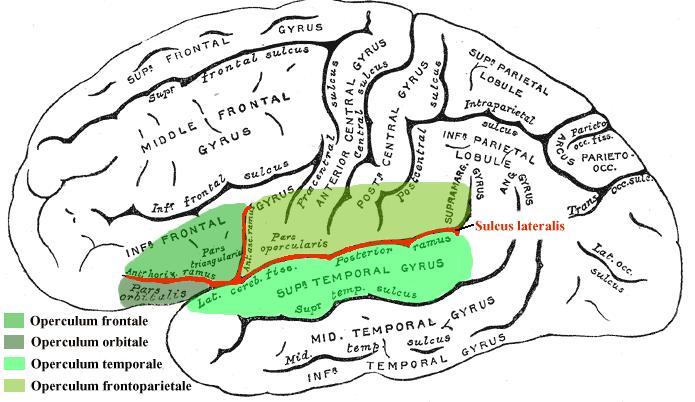
Схематическое изображение покрышки островка (operculum insulae). Этим термином обозначаются прилегающие друг к другу участки лобной, теменной и височной долей, которые «покрывают» находящуюся в глубине латеральной (сильвиевой) борозды островковую долю большого мозга (островок Рейля). В зависимости от принадлежности к той или иной доле выделяют лобную (иногда в её составе упоминают орбитальную), височную и лобно-теменную (или роландическую) покрышки. Именно при поражении последней развивается классический симптомокомплекс Фуа – Шавани – Мари (источник: https://upload.wikimedia.org/wikipedia/commons/b/b3/Operculum.png)
К настоящему времени в литературе описано около 150 случаев. Клиническая картина синдрома обусловлена диссоциацией непроизвольных и произвольных функций жевательной, мимической и бульбарной мускулатуры. Утрачивается возможность выполнения произвольных движений мышц лица, нижней челюсти, языка и глотки. Как следствие этого, больные становятся неспособными говорить, глотать, гримасничать и улыбаться – т.е., выполнять любые произвольные движения мимико-мастикаторной и лингвофарингеальной мускулатуры. В то же время непроизвольные движения указанных мышц сохранены: возможен смех, зевание, кашель, мимическое сопровождение эмоций, закрывание глаз во время сна, мигание.
Достаточно характерным является внешний вид таких больных – рот полуоткрыт, причём, как правило, произвольное закрывание или ещё большее открывание рта невозможны; лицо выглядит амимичным за счёт псевдопериферического двухстороннего пареза мимических мышц, хотя больной может наморщивать брови при взгляде вверх, моргать, полностью закрывать глаза во сне и совершать движения нижней челюстью и лицевой мускулатурой при спонтанных эмоциональных реакциях (смех, плач). Отмечается также псевдобульбарный паралич мышц нёба, глотки и языка с развитием тяжёлой дисфагии, требующей установки назогастрального зонда. При полной или практически полной неподвижности языка никогда не наблюдается фасцикуляций и атрофий. Некоторые больные способны издавать нечленораздельные звуки, но в большинстве случаев имеется анартрия. Способность к общению с помощью письма или жестов в таких случаях сохраняется. К другим проявлениям относятся слюнотечение, сохранные корнеальные рефлексы, появление рефлекса Льюиса – Бивора – Ваттевиля, снижение или исчезновение глоточного рефлекса.
В некоторых случаях к классической картине могут присоединяться афатические расстройства, а также нарушения движений и чувствительности в конечностях за счёт распространения патологического процесса за пределы лобно-теменной покрышки.
Клинически выделяют 5 форм синдрома Фуа – Шавани – Мари: острую форму, обусловленную двухсторонним инфарктом в зоне лобно-теменной покрышки; подострую форму, возникающую при нейроинфекциях, рассеянном склерозе (болезни Шарко – Вульпиана); хроническую прогрессирующую форму, встречающуюся при нейродегенеративных заболеваниях; врождённую форму (двухсторонняя центральная макрогирия или синдром Ворстер-Дроута), обусловленную дефектом миграции нейронов; транзиторную форму у детей с эпилепсией.
Основными отличиями коркового псевдобульбарного синдрома от бульбарного синдрома, поражения черепных нервов или болезни нервно-мышечного синапса (напр., ботулизма или миастении) являются отсутствие нарушения глазодвигательных функций, сохранность или повышение стволовых рефлексов (например, нижнечелюстного рефлекса Бехтерева), диссоциация непроизвольных и произвольных функций заинтересованной мускулатуры, отсутствие атрофий и фасцикуляций мышц. Что касается отличий от псевдобульбарного синдрома внекоркового генеза (субкортикального (капсулостриарного) и понтинного), то характерными для синдрома Фуа – Шавани – Мари являются как правило острое начало, чёткая диссоциация непроизвольных и произвольных функций заинтересованной мускулатуры, отсутствие эмоциональной лабильности, недержания мочи и кала.
Прогноз плохой, особенно в отношении восстановления способности говорить и есть.
Впервые клиническая картина синдрома была описана в 1837г. берлинским врачом A.Magnus (Magnus A. Fall von Aufhebung des Willenseinflusses auf einige Hirnnerven // Müllers Archiv fur Anatomie, Physiologie und wissenschaftliche Medicin, 1837. – S.258–266). Однако, широкой неврологической общественности синдром стал известен после его описания французскими врачами: неврологами Шарлем Фуа и Жаном Альфредом Эмилем Шавани и педиатром Жюльеном Мари в 1926г. (Foix C., Chavany J.A., Marie J. Diplégie facio-linguo-masticatrice d’origine sous-corticale sans paralysie des membres (contribution à l’étude de la localisation des centres de la face du membre supérieur) // Revue neurologique, Paris, 1926. – Vol.33. – P.214-219). Правда, в работе не был описан самый характерный признак синдрома – диссоциация между произвольными и непроизвольными движениями. В последующие годы сообщения об этом синдроме ограничивались франкоязычной неврологической литературой, вплоть до 1980г., когда, с наступлением эры КТ, работа, посвящённая клинико-радиологическому описанию синдрома Фуа – Шавани – Мари, впервые появилась в англоязычной литературе. Врождённая форма была впервые описана английским неврологом Чарлзом Ворстер-Дроутом в 1953г., правда, развитие синдрома автор расценил как последствие перенесённого детьми энцефалита (кстати, после этого в литературе было опубликовано всего лишь несколько сообщений о развитии синдрома Фуа – Шавани – Мари у детей и единичный случай – у взрослого). В последующих своих работах 1956, 1968 и 1974гг. Ворстер-Дроут более подробно описал врождённую форму и связал её с нарушением развития ЦНС. В 1986г. было опубликовано первое клинико-радиологическое исследование этой формы. Транзиторная (функциональная, интермиттирующая) форма синдрома была впервые описана в 1987г. у двух детей с доброкачественной парциальной эпилепсией, проявлением эпилептического статуса у которых явились анартрия, затруднения при глотании и слюнотечение.